
Paired End Sequencing R1 And R2
A) bowtie2 mapping against host genome: Add comment • link modified 17 months ago • written 17 months ago by y.hoogstrate • 460.


Ask question asked 2 years, 4 months ago.
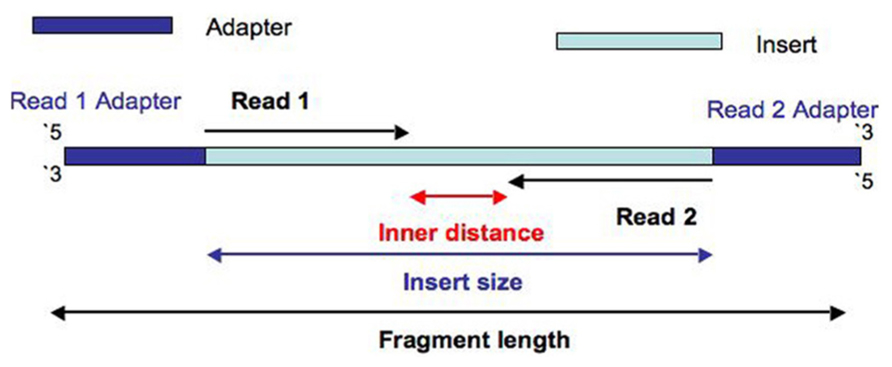
Paired end sequencing r1 and r2. You are only guaranteed that the pairs are complementary. Actually the reads are always mixed just the way you describe them. Read 1 is used to sequence the 16 bp 10x barcode and 10 bp umi, while read 2 is used to sequence the cdna fragment.
R2 = file contains “reverse” reads Each sample index provided in the chromium i7 sample index kit combines 4 different sequences in order to balance across all 4 nucleotides. R1 = file contains “forward” reads.
For the first test, i took some sequence from the human genome (hg19) and created two 100 bp reads from this region. If you have multiples of r1 and r2, you can merge the r1 files together and then r2 files together prior to alignment as long as you ensure files being merged for r1 and r2 are in the same order i.e sequencing run1, run2, run3 fastqs are concatenated in that order for both the final merged r1 and r2 files. Viewed 670 times 3 $\begingroup$ there are commercial sequencing kits/sequencers that allow for paired end sequencing in which the two reads obtained for each fragment are of different length?
Active 2 years, 4 months ago. Due to the way data is reported in these files, special care has to be taken when. Active 2 years, 6 months ago.
We approach the case where r1 and r2 do not overlap and the sequencing of the amplified dna fragment is. Illumina gets sequence data from both strands of input sequence which means it outputs data from both ends of the input and is normally reported two files r1 and r2, often refereed to as mates files (r1=first mates, r2=second mates). Viewed 466 times 2 $\begingroup$ i recently finished mapping an rnaseq run using star2.3.0 and noticed the read1 and read2 counts are different, according to samtools flagstat.
What could cause differing counts of r1 and r2 in paired end sequencing (rnaseq) ask question asked 2 years, 6 months ago. R1 may be forward or reverse. The files have this naming convention:
“r1” forward read and “r2” reverse read. Merging reverse and forward reads 4.16s sequence data from the illumina dna sequencer is produced in the fastq format.
Check to make sure they all have the same number of sequences: 6.for each dna fragment sequenced we have two sequences; So the r1 and r2 reads are split into two different files (one for the data from each end), and it does mean that we have to run two different bwa sessions.
In this case, r1 length is ~60bp, and it. This is different from fr because it means the reverse read aligned at a lower base pair position than the. Depending on your requirements, you may indeed need to check which is which down the pipeline.
Where “xxx” is a file prefix and. The reads have been generally trimmed to remove the low quality reads and adaptor contaminations. But the r1 and r2 files do not have equal number reads, which means some r1 files have more reads and some has less.
If insert size is 150bp, read length usually is ~60bp as quality score after 60th bp is unacceptably low. In the case of 16s rdna hypervariable tag sequencing, these regions are selected by primers targeted to specific regions. Paired end sequencing with r1 and r2 of different length.
Where a pair of reads (r1, r2) overlap, the merging programs assign quality scores for the merged read based on these profiles, and whether the r1 and r2 bases agree (“match”) or not. Write all (mapped and unmapped) reads to a single.bam file. Standard alignment utilities do that automatically.
This would seem to be a suboptimal choice for these. R2 may also be forward or reverse.






{kind=link}
{kind=link}
Posting Komentar untuk "Paired End Sequencing R1 And R2"